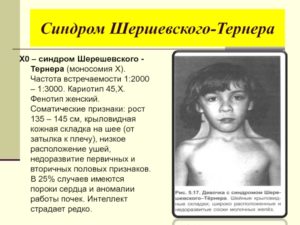
Синдром Шерешевского-Тернера

Синдром Шерешевского-Тернера представляет собой генетическое заболевание, обусловленное отсутствием половой Х-хромосомы или структурными изменениями в ней. Патология в подавляющем большинстве случаев выявляется у девочек, у мальчиков — в крайне редких случаях. Синдром диагностируется примерно у одного из 2500-4500 новорожденных.
Суть патологии заключается в том, что имеет место полная утрата Х-хромосомы или же структурные изменения в ней при нормальном кариотипе, что провоцирует аномалии развития половых желез на ранних этапах развития эмбриона.
Причины развития синдрома
Хромосомные аномалии могут быть обусловлены как генетическим фактором, так и воздействием внешних факторов, таких как ионизирующее излучение или токсические вещества.
Симптомы
В ряде случаев какие-либо характерные симптомы, позволяющие заподозрить синдром Шерешевского-Тернера у новорожденного, могут почти полностью отсутствовать. Однако нередко обнаруживаются внешние специфические признаки, к которым можно отнести:
- Крыловидные складки кожи на шее
- Соски широко расставлены и втянуты
- Отечные конечности (стопы и кисти)
- Низкие показатели роста и веса ребенка при доношенной беременности
В дальнейшем у пациентов с синдромом Шерешевского-Тернера отмечается задержка в росте, что становится все более очевидным уже к подростковому возрасту и составляет у взрослого порядка 25-ти сантиметров.
Среди внешних признаков стоит отметить так называемое «лицо сфинкса» (миопатическое лицо) с вялой мимикой и отсутствием складок на лбу, немного приоткрытым ртом и глазами, которые не полностью закрываются.
В подростковом возрасте у девочек отмечается задержка полового развития, которая характеризуется первичной аменореей, недоразвитием вторичных половых признаков (в частности молочных желез, роста волос на в подмышечных впадинах и на лобке).
Нередко у пациентов с синдромом Шерешевского-Тернера выявляются пороки развития различных органов и систем организма: патологии опорно-двигательного аппарата, сердечно-сосудистой, мочевыделительной системы, органов слуха.
Возможно отставание в интеллектуальном развитии, эмоциональная нестабильность, повышенная тревожность, депрессивные состояния.
Наличие и степень проявления перечисленных симптомов зависят от вариантов заболевания, связанных с характером генетической патологии.
Хотите записаться на прием?
Кариотип при синдроме Шерешевского-Тернера
У пациенток могут определяться различные кариотипы.
Кариотип 45Х
Примерно у половины женщин, страдающих этим синдромом, в результате исследования определяется кариотип 45Х, что свидетельствует об утрате одной хромосомы (в 75% случаев обнаруживается отсутствие хромосомы, наследуемой с отцовской стороны).
Влагалище и матка не обнаруживаются, в то же время в малом тазу присутствуют тяжи, аналогичные тем, которые обнаруживаются при синдроме Рокитанского-Кюстнера. У пациенток с таким кариотипом на месте яичников определяются соединительнотканные тяжи. Ткань яичников полностью отсутствует.
Обследование выявляет повышенный уровень гонадотропных гормонов, в частности ФСГ (фолликулостимулирующего гормона). Выражены внешние характерные признаки, часто имеют место нарушения со стороны различных систем организма.
Мозаичный кариотип
Помимо этого, может быть определен мозаичный кариотип (наличие клеток с различным кариотипом), такой например, как 45Х/46XY или 45Х/46ХХ. В зависимости от мозаичного варианта и количества измененных клеток варьируются проявления синдрома.
Если у женщины определяется кариотип 45Х/46ХХ, то присутствуют яичники, размер которых уменьшен, и матка без аномалий развития.
Для того чтобы оценить репродуктивную функцию пациентки, требуется тщательное обследование в клинике ЭКО, включающее в себя исследование матки, эндометрия и овариального запаса яичников.
У пациенток с таким мозаичным вариантом синдрома Шерешевского-Тернера существует некая вероятность развития беременности без применения методов вспомогательной репродукции. При более значительном снижении фертильности могут быть эффективны различные схемы стимуляции овуляции или ЭКО с донорскими ооцитами.
У пациенток с кариотипом 45Х/46XY преодоление бесплодия требует проведения процедуры суррогатного материнства с использованием яйцеклеток донора, т.к. имеет места аплазия матки и влагалища.
Яичники удаляют, поскольку наличие Y-хромосомы в кариотипе повышает риск развития злокачественных процессов.
При таком мозаичном варианте имеет место вирилизация наружных половых органов (увеличенный клитор, измененный вход во влагалище).
Лечение
Лечение различается в каждом конкретном и случае и может предполагать прием анаболических стероидов (для коррекции отставания в росте) и гормональных препаратов (для развития скелета и вторичных половых признаков), а также проведение пластических операций (для удаления крыловидных складок и коррекции косметических дефектов). Помимо этого, проводится терапия выявленных патологий (удвоение почек, дефект межжелудочковой перегородки, остеопороз и т.д.).
Лечение бесплодия при синдроме Шерешевского-Тернера
Схема лечения бесплодия при синдроме Шерешевского-Тернера, как мы уже говорили выше, разрабатывается лечащим врачом гинекологом-репродуктологом в индивидуальном порядке после тщательного обследования.
Хотите записаться на прием?
Источник: https://nova-clinic.ru/statyi/sindrom-shereshevskogo-ternera/
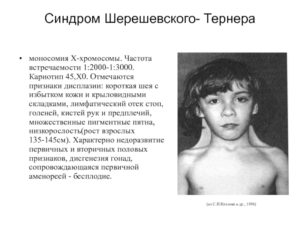
Мозаичная форма синдрома Шерешевского-Тернера
Что это такое, синдром Шерешевского-Тернера? Этот вопрос нередко интересует людей, столкнувшимся с этим заболеванием.
Данная патология являет собой врожденную хромосомную аномалию, которая характеризуется отсутствием одной X-хромосомы (классический вариант).
Учитывая особенности кариотипа, который записывается как 45 XO, очевидным является тот факт, что страдают от соответствующей патологии только представительницы женского пола.
Синдром Шерешевского-Тернера мозаичная форма – патология, при которой не обязательно полностью отсутствует вторая X-хромосома. Чаще она имеет некоторые дефекты, влияющие на общее состояние здоровья пациентки и протекание ее заболевания.
Причины
Причины синдрома Шерешевского-Тернера сокрыты в нарушениях деления клеток и образования генетического материала ребенка еще в период зачатия. Патология фактически возникает в самом начале развития плода.
Нехватка или неправильное распределение целых молекул ДНК ведет к нарушению структуры отдельных хромосом. В данном случае страдает X-хромосома.
При классической форме заболевания она просто отсутствует – 45 XO (в норме 46 XX).
Нехватка генетического материала является причиной развития целого ряда характерных симптомов, которые существенно отражаются на качестве жизни девочки уже с самого рождения.
Однако стоит отметить, что в отличие от некоторых хромосомных аномалий при синдроме Шерешевского-Тернера сохраняется жизнеспособность, а женщины могут вести полноценную жизнь, беременеть и рожать детей.
Факторами, которые способствуют возникновению хромосомной аномалии, являются:
- Половые инфекции во время беременности женщины или в анамнезе.
- Влияние некоторых физических факторов (радиация, электромагнитные поля).
- Влияние химикатов на плод.
- Наследственный фактор.
- Истощение и голодание в период вынашивания плода.
Заболевание может развиваться, даже если перечисленные выше факторы отсутствуют.
Что такое мозаицизм?
Синдром Шерешевского-Тернера мозаичная форма считается более благоприятным заболеванием по сравнению с классической формой, и протекает легче обычного. Причины его возникновения те же, что и указаны выше.
Главным отличием от классической формы болезни является частичное развитие патологии в клетках. В организме одновременно существуют ткани с нормальным кариотипом и аномальным.
Это позволяет компенсировать проявление некоторых симптомов, что делает течение болезни более легким.
В зависимости от характера повреждения второй X-хромосомы, выделяют следующие варианты мозаичной формы заболевания (по кариотипу):
- 46 Х Хр и 46 X Xq.
- 46 Xi(Xq) или 46,X,i(Xp).
- 45 Х/46 XY.
Как видно, практически во всех случаях общее количество хромосом остается нормальным (46). Но, несмотря на этот факт, присутствуют определенные дефекты в плечах одной из них.
Именно данная ситуация является причиной возникновения характерных признаков и симптомов.
Они вызваны преимущественно изменениями в процессе синтеза белков, что, в конечном счете, проявляется различными аномалиями развития.
Клиническая картина
Симптомы синдрома Шерешевского-Тернера достаточно специфичны. Во многих случаях диагноз удается установить визуально при рождении ребенка. Наиболее распространенными признаками патологии являются:
- Недоразвитие половой системы. Оно проявляется отсутствием или слишком маленьким размером яичников, гипоплазией матки и фаллопиевых труб. В пубертатном возрасте нормально не развиваются молочные железы. Соски часто втянуты, широко расположены.Наблюдается снижение нормальных концентраций половых гормонов в крови, что является основной причиной нарушений менструального цикла. Он может вовсе отсутствовать или быть нерегулярным со скудными выделениями. Во многих случаях естественная беременность невозможна.
- Аномалии развития внутренних органов. Данная группа симптомов чаще проявляется стенозом отверстий между камерами сердца, гипоплазией или патологическими изменением структуры почек.
- Другие симптомы. Отмечается низкий рост. Частым признаком болезни является «шея сфинкса» – формирование кожных складок в данной области. Отеки лица, вальгусное искривление локтей, эпикант и деформация грудной клетки также могут выступать признаками синдрома Шерешевского-Тернера.
Учитывая полиморфизм проявлений классической патологии, постановка диагноза редко бывает проблематичной.
Мозаичная форма синдрома Шерешевского-Тернера отмечается более легким протеканием заболевания. Фенотип (внешние проявления) может оставаться нормальным. Единственным обязательным симптомом практически у всех пациенток остается патология развития половой системы. Степень выраженности заболевания зависит от количества пораженных клеток в организме.
Диагностика
Диагностика синдрома Шерешевского-Тернера в большинстве случаев не составляет особого труда, если речь идет о классической форме болезни. Что же касается мозаичной формы патологии, то здесь иногда возникают отдельные трудности. Дело в том, что при нормальном фенотипе клиницисты далеко не всегда могут сразу заподозрить хромосомную патологию.
Лучшим способом диагностики болезни остается кариотипирование. Оно предусматривает анализ хромосомного набора пациентки с установлением наличия дефектов или отсутствия X-хромосомы.
Для верификации болезни у плода проводят пренатальную диагностику. Главными ее этапами являются:
- Выявления потенциальных факторов риска рождения больного ребенка.
- Анализ кариотипа родителей.
- УЗИ плода.
- Кариотипирование малыша еще в утробе.
С помощью этих методов можно установить наличие болезни еще до момента рождения ребенка. Это позволит сразу же начать соответствующую терапию с улучшением качества жизни девочки в будущем.
Осложнения
Осложнения при мозаичной форме синдрома Шерешевского-Тернера в основном связаны с наличием аномалий развития внутренних органов. Пороки сердца могут привести к ранним проблемам с кровообращением. Недоразвитие почек является причиной почечной недостаточности. Расщепление позвоночника приводит к неврологической симптоматике и тому подобное. Все зависит от исходного состояния организма.
Мозаичная форма болезни в этом плане намного благоприятнее. Опасные осложнения практически не возникают. Патология ограничивается снижением качества жизни, что не слишком влияет на ее продолжительность.
Синдром Клайнфельтера: мужчина с женской хромосомой

Елена Шведкина об одном из самых распространенных генетических заболеваний — больные жалуются на бесплодие, эректильную дисфункцию, гинекомастию и остеопороз
Синдром Клайнфельтера — генетическое заболевание, характеризующееся дополнительной женской половой хромосомой Х (одной или даже несколькими) в мужском кариотипе ХY. При этом в мужских половых железах — яичках — образуется недостаточно половых гормонов.
Как известно, генетический набор человека насчитывает 46 хромосом, из которых 22 пары называются соматическими, а 23‑я пара — половая.
Женщины имеют пару половых хромосом ХХ, а мужчины — ХY.
Для синдрома Клайнфельтера обязательно наличие мужской Y-хромосомы, поэтому, несмотря на дополнительные Х-хромосомы, пациенты всегда являются мужчинами.
Классификация: виды кариотипов при синдроме Клайнфельтера
По количеству дополнительных Х-хромосом различают следующие варианты синдрома Клайнфельтера:
- 47,ХХY — наиболее часто встречающийся
- 48,ХХХY
- 49,ХХХХY
Кроме того, к синдрому Клайнфельтера также относят мужские кариотипы, включающие, помимо дополнительных Х-хромосом, дополнительную Y-хромосому — 48,ХХYY. И, наконец, среди пациентов с этим синдромом встречаются лица с мозаичным кариотипом 46,ХY/47,ХХY (то есть часть клеток имеет нормальный хромосомный набор).
История открытия синдрома
Синдром получил свое название в честь Гарри Клайнфельтера — врача, в 1942 году впервые описавшего клиническую картину болезни. Клайнфельтер с коллегами опубликовали отчет об обследовании 9 мужчин, объединенных общими симптомами, такими как слабое оволосение тела, евнухоидный тип телосложения, высокий рост и уменьшенные в размерах яички.
Позднее, в 1956 г., генетики Планкетт и Барр (Е. R. Plankett, М. L. Barr) обнаружили у мужчин с синдромом Клайнфельтера тельца полового хроматина в ядрах клеток слизистой оболочки полости рта, а в 1959 году Полани и Форд (P. E. Polanyi, S. E.
Ford) с сотрудниками показали, что у больных в хромосомном наборе имеется лишняя Х-хромосома.
Активные исследования данной патологии велись в 70‑х годах в США. Тогда всех новорожденных мальчиков подвергали кариотипированию, в результате чего удалось достоверно выявить распространенность и генетические особенности синдрома Клайнфельтера.
Любопытно, что мыши также могут иметь синдром трисомии по половым хромосомам XXY, что позволяет эффективно использовать их в качестве моделей для исследования синдрома Клайнфельтера.
Распространенность заболевания
Синдром Клайнфельтера является одним из наиболее распространенных генетических заболеваний: на каждые 500 новорождённых мальчиков приходится 1 ребёнок с данной патологией.
Кроме того, синдром Клайнфельтера — третья по распространенности эндокринная патология у мужчин (после сахарного диабета и патологии щитовидной железы) и наиболее частая причина врожденного нарушения репродуктивной функции у мужчин.
На сегодняшний день около половины случаев синдрома Клайнфельтера остаются нераспознанными. Часто такие пациенты обращаются за помощью по поводу бесплодия, эректильной дисфункции, гинекомастии, остеопороза, анемии и пр. без установленного ранее диагноза.
Этиология и причины нарушения
Синдром Клайнфельтера относится к генетическим заболеваниям, не передающимся по наследству, поскольку больные, за редким исключением, бесплодны.
Патология, как правило, возникает в результате нарушения расхождения хромосом на ранних стадиях формирования яйцеклеток и сперматозоидов. При этом синдром Клайнфельтера, возникающий за счет нарушения в женских половых клетках, встречается в три раза чаще.
Мозаичные формы обусловлены патологией деления клеток на ранних стадиях эмбриогенеза, поэтому часть клеток у таких пациентов имеет нормальный кариотип.
Причины нерасхождения половых хромосом и нарушения деления клеток на самых ранних стадиях эмбриогенеза до сих пор малоизучены. В отличие от других хромосомных заболеваний, влияние возраста родителей отсутствует или выражено незначительно.
Ранние признаки
В отличие от большинства заболеваний, связанных с нарушением количества хромосом, внутриутробное развитие детей с синдромом Клайнфельтера проходит нормально, склонности к преждевременному прерыванию беременности не наблюдается.
Так что в младенческом и раннем детском возрасте заподозрить патологию практически невозможно. Более того, клинические признаки классического синдрома Клайнфельтера проявляются, как правило, только в подростковом периоде.
Однако есть симптомы, которые позволяют заподозрить наличие синдрома Клайнфельтера в препубертатном периоде:
- высокий рост (пик прибавки роста приходится на период между 5–8 годами);
- длинные ноги (непропорциональное телосложение);
- высокая талия.
У части пациентов наблюдается некоторая задержка в развитии речи.
В подростковом возрасте синдром часто проявляется гинекомастией, которая при данной патологии имеет вид двустороннего симметричного безболезненного увеличения грудных желез. Так как такого рода гинекомастия часто наблюдается у совершенно здоровых подростков, этот симптом часто остается без внимания.
В норме подростковая гинекомастия бесследно исчезает в течение нескольких лет, у пациентов же с синдромом Клайнфельтера обратной инволюции грудных желез не происходит.
В некоторых случаях гинекомастия может не развиваться вовсе, и тогда патология проявляется признаками андрогенной недостаточности уже в постпубертатный период.
Симптомы андрогенной недостаточности при синдроме Клайнфельтера
Андрогенная недостаточность при синдроме Клайнфельтера связана с постепенной атрофией яичек, что приводит к снижению синтеза тестостерона. Степень недостаточности андрогенов резко варьирует.
В первую очередь обращают на себя внимание внешние признаки гипогонадизма:
- скудная растительность на лице или же полное ее отсутствие;
- рост волос на лобке по женскому типу;
- волосы на груди и других частях тела отсутствуют;
- маленький объем яичек (2–4 мл) и их плотная консистенция (патогномоничный признак).
Поскольку дегенерация половых желез, как правило, развивается в постпубертатный период, у большинства пациентов размеры мужских половых органов, за исключением яичек, соответствуют возрастным нормам.
Пациенты могут жаловаться на ослабление либидо и снижение потенции.
У многих мужчин с синдромом Клайнфельтера половое влечение вовсе не возникает, а некоторые — напротив, заводят семью и живут нормальной половой жизнью.
Наиболее постоянный признак патологии — бесплодие, именно оно чаще всего становится причиной обращения таких пациентов к врачу. У 10 % мужчин с азооспемией обнаруживают синдром Клайнфельтера.
Всем пациентам с нарушениями сперматогенеза необходимо определять кариотип для исключения или подтверждения диагноза синдрома Клайнфельтера.
Недостаток андрогенов приводит к развитию остеопороза, анемии и слабости скелетной мускулатуры. У трети больных можно наблюдать варикозное расширение вен голеней.
Андрогены влияют на обмен веществ, поэтому больные с синдромом Клайнфельтера склонны к ожирению, нарушению толерантности к глюкозе и сахарному диабету второго типа.
Доказана предрасположенность таких пациентов к аутоиммунным заболеваниям (ревматоидный артрит, системная красная волчанка, аутоиммунные заболевания щитовидной железы и другие).
Психологические особенности
Коэффициент интеллекта у больных с классическим синдромом Клайнфельтера варьирует от значений ниже среднего до показателей, значительно превышающих средний уровень.
Однако во всех случаях отмечается диспропорция между общим уровнем интеллекта и вербальными способностями, так что нередко пациенты с достаточно высоким IQ испытывают трудности при восприятии больших объемов материала на слух, а также при построении фраз, содержащих сложные грамматические конструкции.
Такие особенности причиняют пациентам много неприятностей в период обучения и нередко продолжают сказываться на профессиональной деятельности.
Данные о психологических особенностях больных с синдромом Клайнфельтера достаточно противоречивы, однако большинство специалистов оценивают пациентов как скромных, робких людей с несколько заниженной самооценкой и повышенной чувствительностью.
Есть данные, свидетельствующие о склонности пациентов с синдромом Клайнфельтера к гомосексуализму, алкоголизму и наркомании.
Сложно сказать, вызваны ли особенности психики у таких больных непосредственным влиянием хромосомной аномалии, или же это реакция на проблемы в сексуальной сфере.
В отношении разных цитогенетических вариантов синдрома Клайнфельтера справедливо правило, что с увеличением количества дополнительных Х-хромосом увеличивается количество и выраженность патологических симптомов.
Диагностика синдрома Клайнфельтера
Во многих странах синдром Клайнфельтера часто диагностируется ещё до рождения ребёнка, так как многие женщины позднего детородного возраста, в связи с высоким риском генетических дефектов у будущего потомства, используют пренатальную генетическую диагностику плода. Нередко пренатальное выявление синдрома Клайнфельтера является поводом для прерывания беременности, в том числе и по рекомендации врачей. В России анализ кариотипа будущего ребёнка проводится крайне редко.
При подозрении на синдром Клайнфельтера проводят лабораторный анализ крови для определения уровня мужских половых гормонов. Необходима дифференциальная диагностика с другими заболеваниями, протекающими с проявлениями андрогенной недостаточности. Точный диагноз синдрома Клайнфельтера ставят на основании изучения кариотипа (набора хромосом) больного.
Исследования, необходимые для подтверждения диагноза
| Анализы | Результаты |
| Кариотип | 47,ХХY (80 % случаев) 48,ХХYY 48,ХХХY 49,ХХХХY 46,ХY/47,ХХY |
| Концентрация ЛГ, ФСГ | Повышена, особенно ФСГ |
| Концентрация общего тестостерона | Чаще снижена (в некоторых случаях нормальная за счет повышения секс-стероид-связывающего глобулина СССГ или на начальной стадии развития заболевания) |
У всех мужчин с резко повышенными концентрациями гонадотропинов необходимо исключить синдром Клайнфельтера, так как нередко первый лабораторный признак этой генетической патологии — повышение в крови концентрации гонадотропинов при нормальном содержании общего тестостерона.
Синдром Клайнфельтера необходимо дифференцировать от других форм первичного гипогонадизма. В любом случае при повышении уровня ФСГ в крови необходимо определение кариотипа для исключения в первую очередь синдрома Клайнфельтера.
Цели лечения синдрома Клайнфельтера:
- Восстановление нормального содержания тестостерона
- Восстановление сексуальной функции
- Ликвидация метаболических нарушений
При клинически выраженной патологии необходима пожизненная заместительная терапия препаратами тестостерона.
Адекватная терапия позволяет не только улучшить внешний вид и общее самочувствие больного, но и вернуть способность к нормальной половой жизни. Кроме того, заместительная терапия предупреждает развитие остеопороза, купирует мышечную слабость. В юном возрасте лечение необходимо начинать сразу же после постановки диагноза.
При синдроме Клайнфельтера лучше использовать препараты тестостерона длительного действия:
- смесь эфиров тестостерона в виде масляного раствора, инъекции которого необходимо делать 2–3 раза в месяц;
- тестостерона ундеканоат в виде масляного раствора — препарат-депо с замедленным высвобождением действующего вещества — инъекции 1 раз в 3 месяца.
Гормонолечение при наличии Х хромосомы у мужчин должно носить постоянный характер. Дозу препарата подбирают индивидуально под контролем уровня тестостерона и ЛГ в сыворотке крови.
Уже развившаяся гинекомастия при синдроме Клайнфельтера не подвергается инволюции даже в случае адекватного лечения, поэтому часто приходится прибегать к хирургической коррекции (мастэктомии).
Для профилактики таких сопутствующих заболеваний, как ожирение и сахарный диабет второго типа, больным рекомендуют придерживаться диеты и следить за собственным весом.
Мониторинг пациентов с синдромом Клайнфельтера следует осуществлять не реже 1 раза в 6–12 месяцев. Он должен включать следующие исследования:
- общий анализ крови для оценки уровня гемоглобина и гематокрита;
- гормональный анализ крови, включающий определение тестостерона и ЛГ (проводится на фоне лекарственной терапии за 1–2 дня до очередной инъекции тестостерона);
- денситометрию (всем пациентам, у которых на момент постановки диагноза были обнаружены остеопения или остеопороз).
Внедрение интрацитоплазматической инъекции сперматозоида в яйцеклетку (ИКСИ) и данные о возможности присутствия зародышевых клеток в яичках у пациентов с синдромом Клайнфельтера предопределили применение метода искусственного оплодотворения для данной категории пациентов, некоторые попытки были удачными.
Прогноз
Прогноз для жизни и трудовой деятельности у пациентов с классическим синдромом Клайнфельтера — в целом благоприятен. Ранняя заместительная терапия, психологическая работа с пациентами и их родителями позволяют больным полностью адаптироваться в современном обществе.
Источник: https://www.katrenstyle.ru/articles/journal/medicine/syndrome/sindrom_klaynfeltera_muzhchina_s_zhenskoy_hromosomoy
Цитогенетическое исследование и молекулярное кариотипирование | Многопрофильная медицинская клиника
Консультация гинеколога-репродуктолога – БЕСПЛАТНО
1. МАТЕРИАЛ ИССЛЕДОВАНИЯ – МАТЕРИАЛ АБОРТУСА.
Собирается при чистке замершей беременности (или анэмбрионии)- это ворсинки хориона, ткани плаценты.
Цель исследования – кариотипирование – исследования хромосом под микроскопом: количество, размеры и морфологические особенности.
Ряд хромосомных заболеваний можно диагностировать по изменению количества хромосом, или по любым другим внутрихромосомным и междухромосомным перестройкам. Любые отклонения от нормы могут стать причиной замирания беременности и аномалий.
Информацию о генетическом коде и возможных мутациях несут ворсинки хориона – ткани плаценты, полученные после выскабливания матки. Если причины остановки развития эмбриона имеют генетическую природу, исследование безошибочно покажет, в какой хромосомной паре возникают нарушения. Важно наличие ворсин хориона в биоматериале.
Только при вакуум-аспирации или выскабливании замершей беременности (анэмбрионии) возможно полноценно взять биоматериал для генетического исследования.
В случае же медикаментозного аборта собрать материал абортуса (сгустки) возможно, но гарантировать наличие ворсин хориона в нем, по которым проводится генетическое исследование, не всегда представляется возможным.
В случае отсутствия ворсин хориона исследование провести невозможно.
Материал собирается строго!!! по инструкции. Методикой сбора биоматериала для генетического исследования владеет хирург, производящий операцию.
Возможны следующие виды генетических исследований материала абортуса:
–цитогенетическое исследование живого клеточного материала (проводится до 14-15 нед. беременности; исследование должно начаться не позже 24 часов после получения биоматериала абортуса с целью сохранения жизнеспособности клетки);
–молекулярно-генетическое исследование по ДНК (срок беременности, как и время проведения исследования значения не имеют).
Методика цитогенетического исследования
Анализ проводится мтодом метафазных пластинок, препараты которых получают из клеток цитотрофобласта ворсинчатого хориона ускоренным “прямым” методом.
Из пробы извлекаются одноядерные лейкоциты и к ним добавляют делящиеся клетки. Исследуются под микроскопом клетки, полученные после остановки этапа деления.
Клетки окрашиваются специальным красителем, фотографируются и исследуются хромосомы под микроскопом.
Показатели нормы: 1. У мужчин – 46 XY 2. У женщин – 46 ХХ
Стоимость цитогенетического исследования в клинике “Медсервис”
| Цитогенетическое исследование материала абортуса, взятого в клинике «Медсервис» | 7 000 руб. | Грубые нарушения, определяемые визуально под микроскопом. В основном анеуплоидии |
| Цитогенетическое исследование материала абортуса, взятого в сторонней организации | 10 000 руб. | #rowspan# |
!!! Важно для сторонних организаций: при сдаче биматериала на цитогенетическое исследование (памятка врачу, как осуществлять забор, прилагается) заполняются анкета и и информированное согласие и вместе с материалом абортуса предоставляются в клинику “Медсервис, соблюдая режим холодильника.
Для цитогенетического исследования важно, чтобы клетки биоматериала были живыми. Чтобы обезопасить себя от невозможности провести исследование на «живом» клеточном материале (его может не быть в материале абортуса по разным причинам) прибегают к методу молекулярного кариотипирования, исследуя ДНК. При этом сохранность клеток в живом состоянии не важна.
Кроме этого сам метод имеет ограничения. Если исследования показывает мужской кариотип, то полученный результат относится к эмбриону, если женский, то результат может относится как к эмбриону, так и матери.
Дальнейшая врачебная тактика заключается в генетическом обследовании матери. При молекулярном генетическом исследовании это исключено, так как исследование идет по ДНК, которая для каждого человека индивидуальна.
Молекулярное кариотипирование материала абортуса
Для проведения исследования используется ДНК, выделенная из биоматериала, а не клеточная культура, как в цитогенетическом исследовании.
Это снижает риск влияния качества биоматериала на возможность проведения анализа (клетки абортивного материала могут не сохранить жизнеспособность внутриутробно и в условиях забора) и исключает в случае женского кариотипа неоднозначность принадлежности исследования матери или эмбриону.
Молекулярное кариотипирование позволяет определить не только грубые нарушения структуры хромосом, видимые под микроскопом, как в случае с цитогенетическим исследованием, но и мелкие субмикроскопические изменения с помощью хромосомного микроматричного анализа. Для этого анализа также берется кровь матери для очищения полученных показателей эмбриона от показателей матери.
Возможно молекулярно-генетическое исследование без предоставления крови матери, тогда исследование будет направлено на выявление анеуплоидий методом секвенирования нового поколения (NGS). Данное исследование выявляет распространенные генетические нарушения кариотипа. Охват остальных патологий генома у данного анализа занимает около 40%, что является ограничением данной методики.
Стоимость молекулярно-генетических исследований в клинике “Медсервис”
| Молекулярное кариотипирование материала абортуса со взятием крови матери | 17000 руб. | выявляет структурные изменения на уровне всего генома, отдельных хромосом, и участков хромосом |
| Молекулярное кариотипирование материала абортуса без взятием крови матери | 10500 руб. | Выявляет анеуплоидии (отсутствие или дополнительные хромосомы) |
!!! Важно для сторонних организаций: при сдаче анализов на молекулярное исследование материала абортуса заполняются анкета и и информированное согласие, берется кровь матери в пробирку К2Е К2EDTA с фиолетовой крышкой и все вместе предоставляется в клинику “Медсервис”, соблюдая режим холодильника.
2. МАТЕРИАЛ ИССЛЕДОВАНИЯ – ВЕНОЗНАЯ КРОВЬ ЧЕЛОВЕКА
Цель исследования – выявление нарушений хромосомного набора человека при планировании беременности. Устанавливается генетическую совместимость партнеров.
Цитогенетическое исследование также необходимо при:
- бесплодии
- наличии у одного из супругов генетических патологий
- невынашиваемости беременности
- нарушении полового развития
- возрастным беременным – старше 35 лет;
- рождении ребенка с наличием генетической патологии
| Определение кариотипа с аберрациями (100 клеток) / 1 пац. | 7950 руб. |
| Определение кариотипа без аберраций | 5100 руб. |
| Определение кариотипа (+ фото) без аберраций / 1 пац. | 6990 руб. |
Позвонить онлайн Записаться на приём
Источник: https://ooomedservis.ru/analizy/citogeneticheskoe-issledovanie/
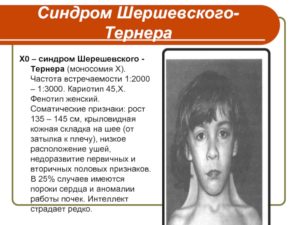
Синдром шерешевского тёрнера : 13 признаков генетического синдрома
25.06.2019
Значительную часть заболеваний человека составляют наследственные патологии, причину которых удалось выяснить медицинской генетике за короткий промежуток времени с начала двадцатого века до наших дней. Некоторые из них сопровождаются бесплодием. Одним из таких заболеваний является синдром Тёрнера-Шерешевского.
Определение понятия
Вся информация о человеке заложена в его генах, которые передаются по наследству из поколения в поколение. Все они разделены на сорок шесть основных составляющих — хромосом. В каждой из них находятся в определённом порядке различные гены.
Все они присутствуют у человека в двойном экземпляре и находятся в сорока четырёх хромосомах. Две оставшиеся обозначаются латинскими буквами X и Y и определяют пол человека.
У женщины в хромосомном наборе две хромосомы Х (46, ХХ), у мужчины одна X и одна Y (46, XY).
Женский пол характеризуется наличием двух Х хромосом, мужской — Х и Y хромосом
При наличии только одной Х хромосомы (45, Х0) развивается заболевание — синдром Тёрнера-Шерешевского, характеризующийся низким ростом, недоразвитием половых органов и множеством аномалий других органов и систем.
Синонимы заболевания: синдром Шерешевского, синдром Тёрнера, синдром Ульриха, синдром Ульриха-Тёрнера.
Впервые заболевание было описано Николаем Шерешевским в 1925 году. Частота заболевания 1 случай на 5 тыс. новорождённых.
Причина синдрома Тёрнера-Шерешевского — отсутствие в наборе одной Х хромосомы
Причины и факторы развития
Пол человека формируется задолго до его рождения. В момент зачатия возможны два сценария:
- Женская половая клетка с набором 23, Х соединяется с мужской клеткой 23, Х с формированием будущего женского организма 46, ХХ. На ранних этапах развития эмбриона человека в утробе одна из Х хромосом инактивируется.
- Женская половая клетка 23, Х соединяется с мужской клеткой 23, Y и определяет развитие эмбриона 46, XY по мужскому типу.
Формирование генома человека в момент зачатия происходит при слиянии половых клеток с гаплоидным (половинным) хромомным набором
Вследствие неправильного формирования половых клеток или их деления на ранних этапах развития эмбриона возможны следующие варианты:
Генетические варианты синдрома Тёрнера-Шерешевского — таблица
Большинство больных синдромом Тёрнера-Шерешевского — женщины. У мужчин заболевание встречается исключительно редко и только в мозаичном варианте.
При простой моносомии все клетки организма содержат одну половую хромосому, поэтому клинические признаки выражены наиболее ярко.
При мозаичном варианте симптомы могут быть сглажены, особенно при небольшом количестве клеток с дефектным хромосомным набором. Возраст родителей не является значимым фактором в формировании синдрома.
Клиническая картина, симптомы и признаки
- У новорождённых с синдромом Тёрнера-Шерешевского наблюдаются следующие симптомы:
- низкий рост и вес при рождении в срок;
- крыловидные кожные образования на короткой шее;
- выраженная отёчность стоп и голеней;
- В возрасте до трёх лет характерны следующие проявления заболевания:
- избыточная двигательная активность;
- плохой аппетит;
- задержка психомоторного развития;
- замедление темпов роста;
- умственная отсталость (в 30% случаев);
- деформация ушей, локтевых суставов, укорочение пястных костей;
- В период полового созревания присоединяется ряд признаков:
- рост ниже среднего (130–145 см);
- широкая грудная клетка;
- частые переломы вследствие разрежения костного вещества;
- искривление позвоночника (сколиоз);
- множественные пигментные пятна на коже (невусы);
- избыточное оволосение;
- неразвитая ткань молочных желёз;
- отсутствие менструации (аменорея);
- У взрослых женщин наблюдается бесплодие (невозможность наступления беременности).
Внешний вид больных с синдром Тёрнера-Шерешевского разных возрастных групп — фото
Крыловидные складки на шее, отек кистей и стоп у новорожденного; внешний вид больной в возрасте 5–6 лет Характерные черты лица и крыловидные складки на шее у ребенка с синдромом Тёрнера-Шерешевского в возрасте 4–5 лет Характерные признаки заболевания: укорочение кисти и низкий рост у пациентки пубертатного возраста Характерные черты лица и крыловидные складки на шее у пациентки зрелого возраста
Выраженность клинической симптоматики зависит от генетического варианта заболевания. При мозаичной форме с небольшим количеством дефектных клеток внешний вид новорождённого не изменяется, болезнь проявляется в период полового созревания.
Диагностика заболевания
Для установки правильного диагноза необходимо проведение следующих мероприятий:
- осмотр врача для выявления внешних признаков заболевания;
- анализ крови на уровень половых гормонов;
- исследование хромосомного набора в клетках, взятых с внутренней поверхности щеки;Анализ кариотипа — основной метод диагности синдрома Тёрнера-Шерешевского
- ультразвуковое исследование органов малого таза проводится для определения размеров матки и яичников, которые обычно значительно уменьшены;
- ультразвуковое исследование сердца для выявления пороков его развития;
- рентгенографическое исследование кистей, позвоночника, локтевых суставов для выявления их деформации и плотности костной ткани;Укорочение кисти за счет костей ладони — характерный признак заболевания
- ультразвуковое исследование почек для выявления аномалий развития;
Дифференциальный диагноз проводится со следующими заболеваниями:
- гипофизарный гипогонадизм;
- синдром Нунан;
- синдром Рокитанского-Кюстнера-Майера;Дифференциальный диагноз заболевания проводится с синдромом Нунан
Гормональная терапия
Основными задачами лечения синдрома Тёрнера-Шерешевского являются достижение приемлемого роста и адекватное течение полового созревания.
Первая решается при помощи назначения гормона роста — Соматотропина вплоть до окончательного закрытия хрящевых зон длинных костей верхних и нижних конечностей.
С 12-летнего возраста для запуска в организме полового созревания, роста молочных желёз и матки, становления менструального цикла назначаются женские половые гормоны — Эстрогены, затем и Прогестерон. Препараты принимаются женщиной с синдромом Тёрнера-Шерешевского в среднем до 50 лет.
Человеческий гормон роста — основной метод лечения синдрома Тёрнера-Шерешевского
Хирургическое лечение
Оперативное лечение проводится в следующих случаях:
- сопутствующий врождённый порок сердца;
- необходимость коррекции деформации позвоночника;
- коррекция крыловидных складок на шее с косметической целью при помощи методов пластической хирургии;
Немедикаментозное лечение
К немедикаментозному лечению синдрома Тёрнера-Шерешевского относятся следующие мероприятия:
- рациональный режим труда и отдыха;
- диета со сниженным количеством углеводов, обогащённая овощами, фруктами и витаминами;
- лечебный массаж;
- лечебная гимнастика;
- электрофорез и магнитотерапия;
- санаторно-курортное лечение;
Народные средства не доказали свою эффективность в борьбе с данным заболеванием.
Прогноз жизни и последствия заболевания
При своевременном установлении диагноза и адекватном проведении лечебных мероприятий прогноз благоприятный. У больных удаётся достичь приемлемого роста и размеров половых органов.
Продолжительность жизни при отсутствии тяжёлых анатомических аномалий со стороны других органов не отличается от таковой у здоровых людей.
Женщина с синдромом Тёрнера-Шерешевского при нормальном размере матки может забеременеть и выносить ребёнка, воспользовавшись современными репродуктивными методами — оплодотворением яйцеклетки, взятой от донора, в пробирке (ЭКО).
Современные репродуктивные технологии — способ родить здорового ребенка для женщины с генетическим синдромом Тёрнера-Шерешевского
Профилактика
Единственным эффективным методом профилактики является дородовая генетическая диагностика с определением хромосомного набора, полученного из околоплодных вод. В последующем проводится консультирование врачом-генетиком.
Синдром Тёрнера-Шерешевского — серьёзное генетическое заболевание, затрагивающее весь организм. При своевременной постановке диагноза пациентки с этим диагнозом могут успешно создать семью и родить здорового ребёнка при помощи современных репродуктивных технологий.
- Елена Тимофеева
- Распечатать
Источник:
Синдром Шерешевского-Тернера
Синдром Шерешевского-Тернера (дисгенезия гонад) – это состояние, при котором наблюдаются нарушения в развитии половых желез. Его провоцирует аномалия половых хромосом.
Синдром Шерешевского-Тернера, согласно с данными медицинской статистики, встречается у одной из четырех тысяч новорожденных девочек и считается одним из наиболее широко распространенных хромосомных аномалий у женщин. Впервые эта патология была описана в 1926 году.
Признаки
Первые признаки нарушения в развитии половых желез наблюдаются уже на ранних стадиях внутриутробного развития эмбриона. В процессе деления половых клеток родителей происходит нарушение в расхождении половых хромосом. Зародыш из-за таких нарушений получает лишь одну Х-хромосому. Как следствие, у него наблюдается неполный набор хромосом.
В результате отсутствия либо изменения половой хромосомы нарушается созревание яичников, что ведет к полному отсутствию нормального полового созревания либо к частичному созреванию. Как следствие, у девочки может развиться бесплодие.
Если у новорожденной отмечается синдром Тернера, то в большинстве случаев наблюдаются только очень легкие признаки патологии.
Но иногда у девочек присутствует выраженная дорсальная лимфедема кистей ступней и рук. Также могут быть складки кожи или лимфедема на задней части шеи.
У больного ребенка отмечается недоразвитие половых органов. У девочки нет яичников. Также недоразвита матка.
Среди аномальных признаков при данном синдроме также могут наблюдаться крыловидные складки шеи. У больных слишком широкая грудная клетка втянутые и широко посаженные соски.
Такие девочки, как правило, имеют более низкий рост, чем их родственники.
Кроме того, симптомами данной патологии иногда являются лимфатические отеки тыла кистей и стоп, аномальная форма ушных раковин, которые часто бывают оттопыренными, а также отмечается тугоухость, медленный рост волос.
Более редкими признаками в данном случае являются птоз, очень низкая линия роста волос на шее сзади, большое количество пигментных невусов, очень короткие четвертые пястная и плюсневая кости, особая форма подушечек пальцев – они выступают и имеют завитки на кончиках пальцев. Также может проявляться гипоплазия ногтей и вальгусное отклонение в локтевом суставе.
Причины синдрома Тернера иногда провоцируют и проявление некоторых аномалий сердца. Это может быть двухстворчатый клапан аорты, коарктация аорты. Как следствие, с возрастом может развиться гипертензия.
Часто встречаются также аномалии развития почек, ЖКТ и гемангиомы. В некоторых случаях заболевание сопровождает косоглазие, дальтонизм, опущение века.
Лечение синдрома Тернера не позволяет избавиться от сопутствующих недугов.
Источник: https://rdbkomi.ru/zabolevaniya/sindrom-shereshevskogo-tyornera-13-priznakov-geneticheskogo-sindroma.html





