Спиноцеребеллярная атаксия: причины, симптомы, диагностика, лечение
Ежедневно человек выполняет большое количество целенаправленных движений, в реализации которых участвуют разные мышечные группы (мышцы-антагонисты, мышцы-синергисты, мышцы-агонисты, мышцы-фиксаторы), вестибулярный аппарат, головной мозг, спинной мозг, зрительный анализатор.
Одним из самых сложных двигательных процессов в нашем организме считается ходьба и поддержание равновесия.
Мышцы туловища и конечностей последовательно сокращаются и расслабляются – появляется шаг, центральная нервная система регулирует скорость шага, инициирует и прекращает ходьбу, адаптирует движение к изменяющимся условиям внешней среды (подъем, спуск и др.), зрительный анализатор передает информацию об окружающем мире, вестибулярный аппарат участвует в поддержании равновесия в пространстве.
В тот момент, когда одна из систем перестает точно контролировать процесс ходьбы или любого другого двигательного акта, появляются дезорганизованные, плохо координируемые движения. Для обозначения такого состояния применяется термин «атаксия».
Атаксия может быть статической, когда человек не может удержать равновесие в стоячем положении, или динамической — нарушается координация движений.
В зависимости от того, в каком образовании расположен очаг поражения, выделяют несколько видов атаксий:
- вестибулярная;
- корковая;
- мозжечковая;
- сенситивная и др.
Спиноцеребеллярная дегенерация
Походка человека с патологией мозжечка неуверенная, шаткая, напоминает походку пьяного.
Термин спиноцеребеллярная дегенерация используется для обозначения группы заболеваний нервной системы, которые проявляются нарушением координации движений в результате сочетанного поражения мозжечка, ствола головного мозга и спинного мозга. В эту группу входят идиопатические и наследственные спиноцеребеллярные атаксии.
Для всех атаксий данной группы общим является прогрессирующее расстройство движений и сочетанное поражение структур ЦНС.
Поражение мозжечка в клинической практике проявляется шаткой походкой (походка пьяного), отклонением туловища в сторону, промахиванием (не может достать пальцем нос, вставить ключ в замок и др.), неверной оценкой расстояния до предмета.
Человек не может быстро выполнить противоположные, чередующиеся действия (повороты рук ладонями вверх и вниз), начинает путаться, сбиваться с ритма. Возникает дрожание в руках или ногах при движении (интенционный тремор). Речь становится отрывистой, замедленной, «рублеными фразами», с неправильной постановкой ударений.
Речевые нарушения обозначают термином мозжечковая дизартрия, а двигательные расстройства – мозжечковая атаксия.
Наследственные спиноцеребеллярные атаксии
Среди всех наследственных заболеваний ЦНС атаксии занимают по частоте второе место после нервно-мышечных заболеваний. На сегодняшний день единая классификация данной группы не разработана, так как наследственные атаксии по клиническим проявлениям очень разнообразны и порой не ясно, это один и тот же вариант или другой.
Остановимся на самых распространенных формах наследственных спиноцеребеллярных дегенераций.
Болезнь Фридрейха
Каждый 120 человек в мире является носителем мутантного гена, который отвечает за развитие болезни. Заболевание передается по аутосомно-рецессивному типу одинаково часто мальчикам и девочкам.
Причины
Механизм развития атаксии связан с нарушением синтеза на митохондриях белка фратаксина, который регулирует транспорт ионов железа через клеточные мембраны. В результате мутации происходит гибель митохондрий и клеток ЦНС, поджелудочной железы, миокарда и других органов.
В ЦНС повреждаются спинно-мозжечковые пути, задние корешки спинного мозга, задние и боковые столбы спинного мозга. На поздних стадиях болезни в патологический процесс вовлекаются ядра черепных нервов, ножки мозжечка, зубчатое ядро, периферические нервы.
Все перечисленные структуры отвечают за координацию движений.
Симптомы
Первые клинические симптомы появляются у детей в возрасте старше 10 лет. Ребенок начинает испытывать трудности при движении в темноте, появляется неуверенная походка, спотыкания и пошатывания.
Постепенно нарушается звукопроизношение (дизартрия), координация движений в руках, которая проявляется затруднением в письме и изменением почерка.
По мере прогрессирования заболевания появляется слабость и атрофия (гибель) мышц ног и рук, которые приводят к полной утрате самостоятельного передвижения и приковывают человека к постели. Тазовые расстройства в виде недержания мочи и кала выявляются у большинства пациентов.
У некоторых больных происходит атрофия зрительных нервов, проявляющаяся полной или частичной утратой зрения. Слабоумие может быть не у всех. На поздних стадиях болезни человек теряет способность к самообслуживанию.
При осмотре у невролога выявляется важный симптом атаксии Фридрейха — исчезновение сухожильных рефлексов (коленные, ахилловы и др.) вплоть до полной арефлексии.
В патологический процесс вовлекается сердце, опорно-двигательный аппарат, эндокринная система. Человек предъявляет жалобы на боли в области сердца, одышку, сердцебиение. Развивается кардиомиопатия. Симптомы со стороны сердечно-сосудистой системы могут появиться намного раньше неврологических проявлений.
Скелетные деформации представлены искривлением позвоночника, «стопой Фридрейха» (высокий свод стопы сочетается переразгибанием пальцев в начальных фалангах и сгибанием их в концевых фалангах). Пальцы рук также поддаются деформации.
Из эндокринных нарушений диагностируют сахарный диабет, дисфункцию яичников, нарушение полового созревания, ожирение и другие заболевания.
Продолжительность жизни при атаксии Фридрайха редко составляет больше 20 лет. Основная причина летального исхода – развитие сердечной или легочной недостаточности, инфекционных осложнений.
Выделяют атипичную форму атаксии, которая характеризуется более поздним дебютом (30–50 лет), благоприятным течением, медленным прогрессированием. При этой форме не выявляют кардиомиопатию, эндокринные нарушения, угасание рефлексов.
Диагностика
- ДНК-тестирование считается решающим методом в диагностике атаксии Фридрейха.
- ЭНМГ (электронейромиография) является одним из важных методов исследования, при котором выявляют поражение чувствительных спинномозговых путей при сохранности двигательных нервов.
- МРТ. Диагностируют атрофию (гибель) вещества спинного мозга.
- ЭХО-КГ, ЭКГ и другие исследования обнаруживают изменения со стороны сердечной мышцы.
- Анализ крови на сахар, гормональный профиль диагностирует эндокринные изменения.
- Рентгенография позвоночника, стоп помогает выявить отклонения.
Диагноз ставится на основании данных всех методов исследования.
Лечение
Специальная терапия не разработана. Все терапевтические мероприятия носят симптоматический характер.
- Лекарственные препараты (никотиновая кислота, рибофлавин, аскорбиновая кислота) улучшают работу митохондрий.
- Физиолечение (электростимуляция).
- Массаж и ЛФК.
- Ортопедические мероприятия: стельки и обувь, операции на позвоночнике.
Наследственная атаксия, обусловленная нехваткой витамина Е
В основе этого заболевания лежит мутация определенного гена, приводящая к дефициту в организме витамина Е.
Заболевание встречается реже атаксии Фридрейха, передается по аутосомно-рецессивному типу. Другое название болезни – «атаксия Фридрейха с дефицитом витамина Е», или «синдром AVED».
Причины
В основе заболевания лежит мутация гена, который отвечает за встраивание витамина Е в структуру липопротеидов низкой плотности (участвуют в антиоксидантной защите клеток).
Симптомы
По клиническим проявлениям атаксию с дефицитом витамина Е практически невозможно отличить от атаксии Фридрейха. Первые симптомы в виде нарушения координации движений появляются у детей в возрасте от 4 лет до 18 лет.
Нарушение речи, угасание рефлексов также встречаются при этой форме. Следует отметить, что поражение сердечно-сосудистой, костной и эндокринной системы встречается в несколько раз реже.
Ближе к 30 годам большинство пациентов теряют способности к самостоятельному передвижению и самообслуживанию, приковываются к постели.
Диагностика
Всем пациентам с атаксиями необходимо проводить анализ крови на определение сывороточного витамина Е. Снижение или отсутствие его концентрации в сыворотке крови является достоверным маркером заболевания.
Лечение
Назначение пожизненной терапии витамином Е в суточной дозировке 5-10 мг\кг приводит к исчезновению симптомов и клиническому выздоровлению человека, особенно при раннем начале лечения.
Аутосомно-доминантные спиноцеребеллярные атаксии
Эта группа атаксий включает в себя ряд самостоятельных заболеваний, которые очень трудно отличить друг от друга по клиническим проявлениям.
https://www.youtube.com/watch?v=ncyRC7dBEfo
Развитие генной инженерии позволило классифицировать их на отдельные единицы с помощью ДНК-диагностики. Сегодня изучено более 13 генов, мутации которых приводят к развитию доминантных атаксий. Заболевания получили названия согласно порядковому номеру гена, на котором обнаружили аномалии: доминантная спиноцеребеллярная атаксия 1 типа, 2 типа, 3 типа,…, 13 типа и т.д.
Причины
Генная мутация приводит к синтезу нерастворимых внутриклеточных молекул, которые вызывают гибель клетки. При каждом типе доминантной атаксии поражаются определенные нервные клетки.
Симптомы
Вне зависимости от типа атаксии, в клинической картине на первый план выступает поражение мозжечка и его связей с другими отделами ЦНС. Выявляют мозжечковую атаксию, мозжечковую дизартрию и другие симптомы.
Доминантные формы атаксий отличаются друг от друга по дебюту симптомов и течению процесса.
- Спиноцеребеллярная атаксия 1 типа возникает в возрасте 30-40 лет. У человека появляются неустойчивые, неловкие движения при быстрой ходьбе и беге. Через несколько лет к симптомам присоединяются нарушение почерка, дизартрия. Походка становится атактической, в руках появляется дрожание при движении. Нарушается координация движений. Одновременно повышается мышечный тонус в руках и ногах, который приводит к снижению полного объема движений в суставах и вынужденному сгибанию конечностей. По мере прогрессирования заболевания, развиваются слабоумие (деменция), тазовые расстройства (недержание кала, мочи), тремор головы, нарушение глотания. Человек полностью утрачивает способности к передвижению. Летальный исход наступает через 10 и более лет от инфекционных осложнений.
- Спиноцеребеллярная атаксия 2 типа по клиническим проявлениям схожа с атаксией 1 типа. Для этого типа характерны согласованные движения глазных яблок (саккады), угнетение сухожильных рефлексов наступает чаще и быстрее.
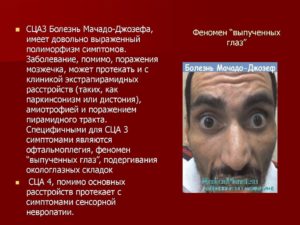
- Спиноцеребеллярная атаксия 3 типа (болезнь Мачадо-Джозефа). Мозжечковые атаксии при этом типе сочетаются с другой неврологической симптоматикой: замедление походки и речи, замедление движений в мимических мышцах, вычурные и насильственные движения в теле и конечностях (дистония), опущение верхнего века, слабость в руках и ногах (парезы), «выпученные глаза», подергивание мышц рта и другие симптомы.
- Спиноцеребеллярная атаксия 4 типа проявляется координаторными нарушениями в сочетании с сенсорной невропатией, которая проявляется болевыми ощущениями и нарушением чувствительности в зоне пораженного нерва.
- Спиноцеребеллярная атаксия 5 и 6 типа. Эти два типа атаксий появляются в возрасте 50-60 лет, медленно прогрессируют, не приводят к глубокой инвалидизации человека, сохраняют способность больного самостоятельно передвигаться и обслуживать себя. Основной симптом – шаткая походка, может быть нарушение речи.
- Спиноцеребеллярная атаксия 7 типа. Характерная особенность данного типа атаксии — сочетание двигательных расстройств с поражением сетчатки глаз, которое может привести к слепоте человека.
- Спиноцеребеллярные атаксии 8 и последующих типов изучены плохо, так как сегодня зарегистрированы единичные случаи этих заболеваний.
Диагностика
- КТ или МРТ (выявляет специфические изменения в головном мозге, исключает другие заболевания).
- ДНК-тестирование (основной метод диагностики).
Лечение
На сегодняшний день специфическая, эффективная терапия отсутствует.
В качестве вспомогательного лечения применяется ЛФК, вестибулярная гимнастика.
Другие формы наследственных спиноцеребеллярных атаксий
В эту группу входят дентаторубропаллидолюисова атрофия (ДРПЛА), эпизодические (периодические, пароксизмальные) атаксии, синдром Маринеску-Шегрена .
Заболевания встречаются крайне редко, лечение только симптоматическое.
Лекция специалиста на тему атаксия Фридрейха:
Источник: https://doctor-neurologist.ru/spinocerebellyarnaya-ataksiya-prichiny-simptomy-diagnostika-lechenie
Как проявляет себя спиноцеребеллярная атаксия? Болезнь Мачадо-Джозефа: что такое, типы, причины, лечение

Группа спиноцеребеллярных атаксий относится к повреждениям мозжечка, которые наследуются по аутосомно-доминантному типу. При этом происходящие в генах мутации провоцируют формирование патологических белковых продуктов, вызывающих отмирание и гибель клеток в мозжечке, спинном мозге, в коре полушарий головного мозга.
Неврологические симптомы спиноцеребеллярной атаксии развиваются довольно медленно, а само по себе развитие может затягиваться на срок до двадцати лет, но может встречать и более быстрое прогрессирование заболевания. Порой наблюдаются периоды стабильного состояния.
При развитии сопутствующих инфекционных поражений начинают проявляться дополнительные симптомы.
Пациенты с сильно запущенными стадиями патологии лишаются возможности вставать с постели, у них формируется дисфагия, то есть нарушение акта глотания, и другие подобные признаки.
Человек может умереть в связи с истощением и часто от развития миокардита, сопровождающегося тяжелыми формами недостаточности сердца. При надлежащем уходе больной доживает до сорока — пятидесяти лет.
Это важно! Спиноцеребеллярная атаксия характеризуется большим количеством проявлений, которые группируются в зависимости от конкретного случая болезни.
Первоначально симптоматика проявляется практически незаметно — появляется неловкость в движениях, неустойчивость во время быстрой ходьбы, при беге.
Постепенно симптомы всё больше активизируются и провоцируют типичную для болезни шатающуюся походку.
Впоследствии симптоматика дополняется тремором ног и рук при реализации каких-либо действий и при нарушении координации движений. Кроме того, начинается изменение почерка — он становится неровным, а буквы слишком большими, также изменяется речь.
Для заболевания характерны и расстройства движения глаз — толчкообразные, резкие движения глазных яблок при смещении взгляда с одного объекта на другой.
Часто отмечается возникновение нарушений глотания, произношения речи, нарушения работы слухового аппарата, нарушение стула и отхождения мочи, паралич ног и рук, развиваются патологические рефлексы.
А привычные рефлексы одновременно ухудшают реакции и затем исчезают полностью.
Временами заболевания протекает в абортивной или легкой форме и провоцирует незначительную инвалидность пациента или вовсе не сопровождается инвалидностью. Такие патологии могут диагностироваться у родственников больного, которые страдают развернутыми клиническими типами болезни.
Врачи отмечают прогрессирующую потерю мышечной массы, сопровождающуюся слабостью пораженных мышц, кратковременное произвольное сокращение сразу нескольких волокон мышц, а это проявляется трепетанием под кожным покровом.
Это важно! Часто церебральная атаксия характеризуется непроизвольными передвижениями головой и конечностями, поэтому развивается слабоумие. Реже отмечаются симптомы заболевания Паркинсона. Также могут возникать патологические изменения зрительных органов — ухудшение или полная потеря зрения.
При проведении МРТ или компьютерной томографии для головного мозга диагностируется уменьшение размера мозжечка. Болезнь активно прогрессирует и поэтому приводит к инвалидности. В основном причиной летального исхода становятся инфекционные осложнения.
Лечение
Хотя атаксия обычно не излечима, многое можно сделать, чтобы облегчить симптомы, улучшить качество жизни пациента. Некоторых люди восстанавливаются спонтанно, другие испытывают дополнительные осложнения.
Адаптивные устройства, такие как трость или трость, ходунки, инвалидные коляски или костыли, используются для оказания помощи пациентам в координации и балансировании, чтобы помочь достичь максимальной независимости.
Симптомы, такие как тремор, скованность, спастичность, нарушения сна, мышечная слабость, депрессия (грусть, гнев) устраняются при помощи целенаправленной физической терапии, логопедии, медикаментов, психологического консультирования.
- Профессиональная терапия: помогает человеку лучше справляться с работой дома, помогает сделать кухню более практичной.
- Речевая терапия: помогает при проблемах глотания, кашле, удушье, речи. Логопед также помогает пациенту научиться пользоваться средствами речи, если речь становится очень сложной.
- Ортопедическая помощь: для лечения искривления позвоночника.
- Физическая терапия, физиотерапевт: для сохранения силы, улучшения мобильности.
- Психологическое консультирование: занятия нужны, чтобы справиться с депрессией, которая возникает, когда симптомы влияют на физическую мобильность и координацию.
- Диета, добавки, питание: Одной из причин атаксии является отсутствие витамина Е или В12. Пациенты с низким уровнем этих витаминов должны принимать добавки и иметь специальную диету. Некоторые могут быть чувствительны к глютену, поэтому назначается диета без глютена.
- Лекарства, медикаменты: Лекарственные средства не обязательно потребуются для лечения вестибулярной атаксии, но нужны для снятия симптомов, таких как головокружение. Лечение telangiectasia производится инъекциями гамма-глобулина для усиления иммунной системы. Существуют также препараты для устранения мышечных спазмов и неконтролируемых движений глаз.
Наследственная атаксия Пьера Мари
Мозжечковая атаксия Мари – это генетический недуг, который передается из поколения в поколение. Вероятность избежать этой болезни крайне мала, если кто-либо из близких кровных родственников (мать или отец) страдал ею. Случаи, когда один из родителей болел синдромом Пьера Мари и не передал этот генотип ребенку, почти не встречаются в медицинской практике.
Наследственная атаксия начинает впервые проявляться в возрасте 30-35 лет: тогда наблюдаются нарушение координации движений и походки. После прибавляются проблемы с речью и потеря мышечного тонуса верхних конечностей.
Синдром Пьера Мари может привести к полному слабоумию к 50-55 годам, если вовремя не принять соответствующие меры профилактики. Поэтому, если вы знаете, что у вас может развиться наследственная мозжечковая атаксия, обращайтесь к врачу до 30-ти лет.
Дополнительные симптомы
По мере развития спиноцеребеллярная атаксия обрастает новыми симптомами:
- возникает тремор конечностей после каких-либо действий или в результате нарушения координации;
- сильно меняется почерк – буквы становятся неровными, большими;
- постепенно изменяется речь, но этот признак сложно заметить, если жить с пациентом постоянно;
- возникают расстройства глазных движений – они становятся резкими, толчкообразными, когда человек перемещает взгляд с объекта на объект;
- в некоторых случаях ухудшается работа слухового аппарата;
- у многих людей нарушается стул, мочеиспускание;
- в запущенных случаях появляется паралич рук и ног;
- также заметно ухудшение привычных реакций и рефлексов, со временем они могут полностью исчезнуть.
У некоторых больных спиноцеребеллярная атаксия протекает в легкой форме, иногда переходит в стадию с присвоением временной инвалидности, но чаще всего не требует ее. Подобная картина встречается у людей, чьи близкие родственники страдают спиноцеребеллярной атаксией развернутого плана.
При спиноцеребеллярной атаксии всегда наблюдается потеря мышечной массы, которая в результате приводит к судорогам, выраженной мышечной слабости. Один из симптомов – ощущение подергивания, трепета под кожей.
Иногда при спиноцеребеллярном поражении возникают непроизвольные повороты и подергивания головой, развивается слабоумие, признаки которого похожи на болезнь Паркинсона. Для нарушения характерны и проблемы со зрением. Тяжелая форма сопровождается полной утратой зрительного контакта.
Виды спиноцеребеллярной дегенерации
Несмотря на свою наследственную природу, данный вид дегенераций начинает проявляться чаще всего не ранее, чем в 20-летнем возрасте. Первые проявления обычно наблюдаются между 20 и 60 годами, хотя некоторые виды этих атаксий могут дебютировать и в первые годы жизни.
Принято выделять как минимум 7 типов спиноцеребеллярных дегенераций. Некоторые из них получили названия по имени ученых, описавших их особенности и признаки. Каждый из этих видов отличается теми или иными проявлениями в большей или меньшей степени, возрастом пациентов, накоплением или дефицитом некоторых веществ, скоростью развития болезненных процессов.
Наиболее часто встречающимся типом является атаксия Фридрейха. Это заболевание проявляет себя чаще в детском возрасте (от 2 до 15 лет). Главной причиной проблем в организме является мутация гена, кодирующего белок фратаксин.
В результате он не вырабатывается в нужном количестве, из-за чего возникает избыток в организме железа и свободных радикалов, что и приводит к повреждениям нервной системы и различных органов.
Довольно часто при атаксии Фридрейха развиваются эндокринные нарушения, в частности сахарный диабет. Это приносит пациенту дополнительные проблемы.
Клинические варианты болезни Мачадо-Джозефа
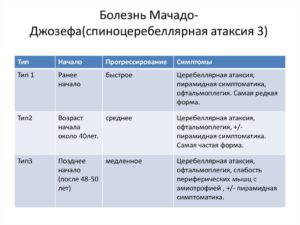
Болезнь Мачадо-Джозефа I тип манифестирует в возрастном периоде 10-30 лет. Характеризуется сочетанием пирамидных и экстрапирамидных симптомов.
Пирамидный синдром (поражение кортикоспинальных трактов) обычно дебютирует спастическим парапарезом, затем присоединяется слабость в руках, парез мышц глотки с развитием дисфагии и дизартрии, парез глазодвигательных нервов с офтальмоплегией (симптом «фиксированных глазных яблок»). Наблюдается клонус стоп, патологические рефлексы.
Экстрапирамидный синдром проявляется симптомами торсионной дистонии, атетозом, вторичным паркинсонизмом. Формируется скованная медленная походка с широкой расстановкой ног. Отмечается шаткость при ходьбе, обусловленная спастикой мышц, а не атаксией.
Типичен экзофтальм, крупные фасцикулярные сокращения языка, не сопровождающиеся его атрофией. Возможны фасцикуляции мимических мышц, миокимия век. Наблюдается вертикальный и горизонтальный нистагм, саккады (однонаправленные движения глаз) с повышенной/пониженной амплитудой.
Источник: https://bolezn.info/nevralgiya/kak-proyavlyaet-sebya-spinotserebellyarnaya-ataksiya-bolezn-machado-dzhozefa-chto-takoe-tipy-prichiny-lechenie.html
Болезнь Мачадо-Джозефа
Болезнь Мачадо-Джозефа, которую также называют спиноцеребеллярной атаксией III типа, является редкой, наследственной атаксией (отсутствие мышечного контроля), которая оказывает свое влияние на центральную нервную систему и характеризуется медленным вырождением заднего мозга. Пациентов, с болезнью Мачадо-Джозефа, со временем может парализовать, но их интеллект остается неизменным. Дебют болезни варьируется, от раннего подросткового и до пожилого возраста.
Эта болезнь имеет три формы. Отличаются эти типы возрастом в котором дебютирует болезнь и степенью тяжести. Болезнь с раним началом приводит к более серьезным последствиям.
Болезнь Мачадо-Джозефа. Эпидемиология
Болезнь Мачадо-Джозефа является редким наследственным неврологическим расстройством, оно чаще всего встречается у лиц португальского происхождения, особенно у выходцев из Азорских островов и островов колонизированных португальцами. Болезнь Мачадо-Джозефа незначительно чаще развивается у лиц мужского пола.
Болезнь Мачадо-Джозефа. Причины
Ген, мутации в котором несут ответственность за развитие болезни Мачадо-Джозефа, расположен в локусе 14q24.3-q31. У пациентов с этой болезнью идентифицируется аномальное количество CAG повторов (тринуклеотидные триплеты) в ДНК гена.
Нормальная последовательность ДНК гена (у здорового человека), как правило, содержит в себе от 12 до 43 копий CAG тринуклеотидов, а у лиц с болезнью Мачадо-Джозефа таких копий может быть от 56 до 86. Тяжесть симптомов / проявлений и возраст в котором начинает развиваться эта болезнь непосредственно связаны с количеством этих повторов.
Таким образом у лиц с первым типом этой болезни будет меньше всего таких аномальных триплетов, а у лиц с третьим типом будет самое большее их количество.
Болезнь Мачадо-Джозефа. Похожие расстройства
- Болезнь Галлервордена-Шпатца – редкое наследственное заболевание, характеризующееся неврологической дегенерацией. Симптомы и проявления этой болезни могут включать в себя: медленная и стабильная мышечная атрофия в руках, ногах, лице, шее, в ротовой полости или спине, мышечные спазмы, невнятная речь (дизартрия), умственная отсталость, нарушение речи (дисплазия).
- Оливопонтоцеребеллярная атрофия – группа редких наследственных неврологических расстройств, характеризующихся прогрессирующей неврологической дегенерацией. Эти заболевания затрагивают внешние слои мозжечка, что в конечном счете приводит к нарушениям в координации движений (атаксия). Симптомы и проявления варьируются и могут включать в себя: мышечные спазмы, непроизвольные движения, аномальные позы, невнятная речь (дизартрия) и экстрапирамидные знаки.
- Прогрессивный супрануклеарный паралич – редкое неврологическое расстройство, оно характеризуется спастической слабостью мышц, контролируемых черепными нервами (например, лица, горла и языка). Симптомы и проявления могут включать в себя: потеря равновесия при ходьбе, жесткая походка (атаксия) или необъяснимые падения.
- Боковой амиотрофический склероз – редкое заболевание нервов скелетных мышц. Болезнь чаще всего затрагивает верхнюю и нижнюю части тела, что в результате приводит к мышечной слабости и постепенной атрофии мышц. Ранние симптомы и проявления бокового амиотрофического склероза могут включать в себя: мышечная слабость, неуклюжие движения рук и трудности в выполнении задач, требующих тонких и точных движений пальцев и рук.
- Атаксия Фридрейха — редкое наследственное заболевание, характеризующееся дегенеративными изменениями и прогрессирующим ухудшением головного и спинного мозга. Симптомы и проявления этой атаксии включают: мышечная слабость и онемение в руках и ногах, искривление позвоночника и паралич ног.
- Атаксия Мари – редкое наследственное заболевание, характеризующееся неврологической прогрессирующей потерей мышечной координации и неустойчивой походкой (атаксия). Прогрессивная дегенерация спинного нерва приводит к потере мышечной массы (амиотрофия) в руках, ногах, голове, шее.
Болезнь Мачадо-Джозефа. Симптомы и проявления
Симптомы и проявления болезни Мачадо-Джозефа I типа дебютируют в возрасте от 10 до 30 лет. Прогрессирование этой болезни, как правило, достаточно быстрое.
Признаки болезни могут включать в себя: сильная слабость в руках и ногах (дистония), спастичность или мышечная жесткость, атаксия, часто в сочетании с шаткой походкой, которая может быть ошибочно принята в качестве проявления алкогольного опьянения, невнятная речь, повреждение мышц контролирующих движения глаз (офтальмоплегия) и выпуклые глаза (экзофтальм). Умственная отсталость не встречалась.
Симптомы и проявления болезни Мачадо-Джозефа II типа аналогичны первому типу, но скорость прогрессирования более медленная.
Клинические признаки болезни II типа, как правило, дебютируют в возрасте от 20 до 50 лет.
Отличительной характеристикой II типа является прогрессирование дисфункций мозжечка, что приводит к развитию шаткой походки (атаксия), к координационным сложностям и к спастическим мышечным движениям.
Болезнь Мачадо-Джозефа III типа начинает проявляться позже, между 40 и 70 годами.
Этот тип характеризуется шаткой походкой (атаксия) и отличается от других форм потерей мышечной массы (амиотрофия), которая развивается из-за воспаления и дегенерации периферических нервов.
Потеря чувств, потеря болевой чувствительности, ухудшение координации рук / ног и сахарный диабет, являются самыми частыми признаками болезни Мачадо-Джозефа III типа. Прогрессирование этого типа самое медленное.
Поставить пациенту правильный диагноз достаточно тяжело, так как определенные симптомы и проявления могут напоминать те, которые развиваются на фоне других неврологических расстройств, таких как болезнь Паркинсона или рассеянный склероз.
Болезнь Мачадо-Джозефа. Диагностика
Золотым стандартом в диагностике этой болезни является прямое определением количества подозрительных триплетов CAG в ДНК гена. Этим методом диагностируется почти 100% случаев. Этот тест выполняется в большинстве специализированных генетических лабораториях.
Болезнь Мачадо-Джозефа. Лечение
Лечение болезни Мачадо-Джозефа только симптоматическое и поддерживающее.
Источник: http://redkie-bolezni.com/bolezn-machado-dzhozefa/
Болезнь Мачадо-Джозефа: что такое, типы, причины, лечение

Болезнь Мачадо-Джозефа (также называемая спиноцеребеллярная атаксия 3 типа) — является одной из примерно 30 признанных, преимущественно наследственных форм атаксии.
Атаксия — это общий термин, который означает отсутствие контроля или координации мышц.
Болезнь Мачадо-Джозефа характеризуется медленными прогрессивными нарушениями движений рук и ног, нарушениями равновесия при ходьбе, что может быть ошибочно принято за состояние опьянения, трудности с речью и глотанием, нарушениями движений глаз, в отдельных случаях сопровождающимися раздвоенным зрением или выпученными глазами, и спастичностью нижних конечностей. У некоторых людей развивается дистония (долговременные мышечные сокращения, приводящие к скручиванию тела и конечностей) повторяющиеся движения и аномальные позы или симптомы, сходные с симптомами болезни Паркинсона. У других больных могут развиться подергивания лица или подергивания языка, невропатия или проблемы с мочеиспусканием и нарушения функционирования вегетативной нервной системы.
Клинические проявления болезни могут быть очень изменчивыми, даже среди больных являющихся родственниками. Такой широкий диапазон симптомов отражает специфический тип мутации, являющаяся причиной болезни: повторное расширение кода ДНК, который различается по размеру у больных лиц. Чем дольше длится экспансия, тем наблюдается более тяжелая форма болезни.
Другими словами, более длительные повторные экспансии, как правило, вызывают заболевание, которое начинается в раннем периоде жизни и показывает более широкий спектр неврологических симптомов.
У большинства людей, страдающих болезнью Мачадо-Джозефа, симптомы как правило возникают в третьем-пятом десятилетии жизни, но могут начинаться уже в раннем детстве или в возрасте до 70 лет.
Болезнь Мачадо-Джозефа — прогрессирующее заболевание, это означает, что симптомы со временем ухудшаются.
Ожидаемая продолжительность жизни варьируется от середины 30-х годов для больных с наиболее тяжелыми формами раннего начала болезни до почти нормальной продолжительности жизни у больных с умеренной формой болезни. Причиной смерти больных с ранним летальным исходом, часто является аспирационная пневмония.
Название «Мачадо-Джозефа» происходит от двух семей португальского / азорского происхождения, которые были среди первых семей, описанных с уникальными симптомами болезни в 1970-х годах.
Распространенность заболевания наиболее высока среди людей португальского / азорского происхождения.
Например, среди иммигрантов с португальскими корнями в Новой Англии распространенность составляет около 4 000 человек, а самая высокая распространенность в мире, примерно одна в 140, происходит на небольшом азорском острове Флорес.
Вскоре после обнаружения дефекта гена была обнаружена наследственная атаксия в европейских семействах, известная как спиноцеребеллярная атаксия 3 типа, с той же самой мутацией. Таким образом, спиноцеребеллярная атаксия 3 типа и болезнь Мачадо-Джозефа являются одним и тем же заболеванием.
Типы болезни Мачадо-Джозефа
Все люди с MJD имеют ту же мутацию гена болезни: повторное расширение ДНК в гене ATXN3. Широкий диапазон симптомов среди пострадавших лиц заставлял исследователей отделить болезнь от разных типов, которые широко различаются по возрастному началу и диапазону симптомов.
Тип I MJD характеризуется началом от 10 до 30 лет, с более быстрой прогрессией и большей дистонией и жесткостью, чем атаксия. Тип II, наиболее распространенный тип MJD, Все больные, страдающие болезнью Мачадо-Джозефа имеют одну и ту же мутацию гена болезни: повторное расширение ДНК в гене ATXN3.
Широкий диапазон симптомов среди пострадавших лиц вынуждал ученых выделить разные типы болезни, которые широко отличаются по возрастному началу и диапазону симптомов.
Болезнь Мачадо-Джозефа 1 типа характеризуется развитием в возрасте от 10 до 30 лет, с более стремительной прогрессией и большей дистонией и жесткостью, чем при атаксии.
Болезнь Мачадо-Джозефа 2 типа — наиболее распространенный тип данного заболевания, как правило начинается в возрасте от 20 до 50 лет, имеет среднюю скорость прогрессирования и вызывает различные симптомы, которые включают заметную атаксию, спастическую походку и усиленные рефлекторные реакции.
У больных, страдающих болезнью Мачадо-Джозефа 3 типа, наблюдается самое позднее начало заболевания (начиная примерно с 40 до 70 лет), которое прогрессирует относительно медленно и характеризуется так же периферическим нервно-мышечным вовлечением (мышечное подергивание, слабость, атрофия и аномальная чувствительность, онемение, покалывание, судороги и боль в руках и ногах), как при атаксии. Большинство больных, особенно с типами I и II, испытывают проблемы со зрением, а именно, раздвоение зрения или помутнение в глазах, утрату способности различать цвета и / или контрасты, а также неспособность контролировать движения глаз. У некоторых больных также наблюдаются заметные симптомы болезни Паркинсона, такие как медлительность движений, жесткость мышц или жесткость конечностей и туловища, а также тремор (дрожь) в руках.
Причины болезни Мачадо-Джозефа
Болезнь Мачадо-Джозефа классифицируется как одна из многих преимущественно наследственных атаксий, а именно спиноцеребеллярных атаксий (SCA).
Известно порядка 30 отдельных генетических причин заболевания, которые приводят к дегенерации клеток в заднем мозге, что приводит к нарушению координации движения.
Задний мозг включает в себя мозжечок (большой отдел мозговой ткани, расположенной в задней части головы), ствол головного мозга и верхнюю часть спинного мозга.
Болезнь Мачадо-Джозефа наследуется по аутосомно-доминантному типу, а это означает, что у больного имеется один вызывающий болезнь аллель генов (аллель — несколько генов, расположенных в одной и той же позиции на хромосомах человека) и нормальный аллель, который может передаваться следующему поколению.
У любого ребенка подверженного болезни родителя с 50% вероятностью имеется риск наследования аллеля болезни. Если ребенок наследует болезнетворный ген, у него в конечном итоге развиваются симптомы заболевания.
У ребенка, который не наследует аллель болезни, не развивается болезнь и он не может передать ее следующему поколению.
Болезнь Мачадо-Джозефа относится к классу генетических заболеваний,при которых происходят мутации в в генетическом коде ДНК, а именно аномально длинные повторения обычного повторения трех букв генетического кода.
В случае данной болезни кодовая последовательность «CAG» повторяется в гене ATXN3, который продуцирует белок болезни, называемый атаксин-3. Когда данный белок мутирует, он подвержен неправильному скапливанию в пораженных клетках головного мозга.
Накопленный белок ataxin-3 образует аномальные агрегаты, называемые тельца включения, которые расположены в ядре клетки.
Одна необычная особенность данного заболевания и многих других распространенных генетических заболеваний — это странный факт, что дети больных родителей склонны к развитию симптомов заболевания ранее в жизни и могут испытывать более тяжелые симптомы.
Это связано с тем, что расширение кода повторяющейся мутации еще больше расширяется при передаче ее следующему поколению, особенно когда она передается мальчику.
Поскольку более длительные экспансии, как правило, вызывают более раннее и более тяжелое заболевание, этот молекулярный рост от одного поколения к другому также вызывает, в среднем, более ранний возраст начала в последующих поколениях.
Хотя более долгие повторы, как правило, вызывают заболевание с ранним началом, невозможно точно предсказать время и ход заболевания для человека основываясь исключительно на длине повторения кода ДНК.
По всему миру болезнь Мачадо-Джозефа, являются наиболее распространенной аутосомно-доминантной унаследованной формой атаксии.
Лечение болезни Мачадо-Джозефа
Болезнь неизлечима, но некоторые симптомы заболевания поддаются контролю.
Лекарственная терапия леводопой (используется при лечении людей, страдающих болезнью Паркинсона) позволяет облегчить «паркинсонические» симптомы (жесткость и медлительность движений, часто сопровождающихся тремором) в течение длительного времени.
Антиспазматические препараты, такие как баклофен, могут помочь уменьшить спастичность. Ботулинический токсин позволяет лечить сильную спастичность и некоторые симптомы дистонии, но ее следует использовать в качестве крайней меры из-за возможных побочных эффектов, таких как трудности при глотании (дисфагия).
Проблемы речи (дизартрия) и дисфагия могут лечиться с помощью лекарственных препаратов и терапией речи. Ношение призматических очков может улучшить размытость или раздвоение зрения. Глазная хирургия имеет только краткосрочные преимущества в результате прогрессирующей дегенерации глазных мышц.
Физиотерапия используется для помощи преодоления проблем при ходьбе, а физические приспособления, такие как ходунки и инвалидные коляски, могут помогать больным в повседневной деятельности. Дневную сонливость, распространенную жалобу при болезни Мачадо-Джозефа (как и нарушение сна в целом), можно контролировать с помощью модафанила.
Другие симптомы, такие как судороги и дисфункция мочи, необходимо контролировать с помощью лекарственных средств и медицинской помощи.
ЛАБОРАТОРНЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ ДЕТЕЙ ПЕРВЫХ 3 ЛЕТ ЖИЗНИ В НЕВРОЛОГИЧЕСКОЙ КЛИНИКЕНА ГЛАВНУЮ СТРАНИЦУ
Источник: http://nevrologvolgograd.ru/bolezn_machado_dzhozefa_chto_takoe_tipyi_prichinyi_lechenie.html
Причины
В тот период, когда патология была впервые обнаружена, специалисты не могли дать однозначного ответа на вопрос о том, каковы причины ее возникновения. Даже несмотря на то, что было выдвинуто множество предположений, ни одно из них не нашло своего научного подтверждения.
Однако, с течением времени и по мере развития ДНК-исследований стало ясно, что главными причинами болезни выступает мутация генов, передаваемая по наследству аутосомно-доминантным путем.
Таким образом удалось выяснить, что заболевание имеет наследственный характер. Отклонения от нормы наблюдаются в 14 хромосоме и сводятся к экспансии комбинации «цитозин-аденин-гуанин».
Количество повторов триплета ЦАГ очень сильно различается, в среднем показатели держатся в диапазоне от 62 до 84. Для сравнения: в норме эта цифра не должна быть выше 37.
Чем данный показатель выше, тем раньше возникает болезнь у человека.
У больных возникает апоптоз нейронов в коре головного мозга, дегенеративные процессы красного и зубчатого ядер, моторных ядер ЧМ-нервов.
Поражение зон мозга приводит к возникновению целого ряда признаков, которые по мере развития патологии и отсутствия соответствующего лечения начинают тревожить больного.
Специалисты сходятся во мнении, что от момента возникновения патологии до проявления первых ее симптомов проходит от 3 до 6 месяцев, но многое зависит также от индивидуальных особенностей организма – у одних больных патология может проявиться значительно раньше, у других – позже.
Клинические варианты патологии
Патология первого типа возникает в возрастном диапазоне 10-30 лет. У больных ярко выражены экстрапирамидные и пирамидные симптомы развития патологии.
Пирамидный синдром возникает вместе с спастическим парапарезом, постепенно появляется значительная слабость в верхних конечностях, парез мышц глотки, глазодвигательных нервов, в результате чего человек теряет способность передвигать глазными яблоками.
Со временем развиваются патологические рефлексы, клонус стоп. Больные начинают испытывать трудности при передвижении, походка становится неуклюжей, скованной, при этом человек ходит с широко расставленными ногами.
Очень часто такие пациенты сильно шатаются во время ходьбы. Нередко у больных смещаются глазные яблоки вперед, возникают значительные фасцикулярные сокращения языка, мимических мышц, глазных век.
Имеют место быть непроизвольные движения глаз высокой частоты.
Патология второго типа возникает чаще всего с 20 до 40 лет. Симптомы заболевания проявляются в мозжечковой атаксии: возникает дизартрия, больной не способен сохранять равновесие, появляется абазия. Характерной комбинации мозжечковых и пирамидных признаков, но второй тип имеет некую схожесть с первым типом болезни.
Патология третьего типа выражается в комбинации амиотрофий и мозжечковой атаксии. Данный тип заболевания возникает довольно поздно – как правило, только после 40 лет.
У больных появляется мышечная атрофия, которая проявляется сильной слабостью и полной потерей сухожильных рефлексов. Особенностью данного типа болезни является наличие всех видов расстройств чувствительности по дистальному типу.
Дегенеративные изменения в кортикоспинальных трактах и поражение экстрапирамидной системы не наблюдается.
Диагностика
Диагностика болезни осложняется тем, что ей свойственна огромная вариабельность повторов триплета ЦАГ, в результате чего болезнь проявляется в самых разнообразных симптомах даже в том случае, если возникает у членов одной семьи. Очень часто болезнь поражает разные поколения близких родственников, причем не обязательно проявляется в одном и том же типе.
Известны случаи, когда только одно из проявлений позволяло заподозрить наличие болезни у разных людей, имеющих между собой родственную связь. Одним из важнейших моментов в диагностике заболевания является консультация генетика, который осуществляет исследование генеалогического древа, учитывая большое количество родственников, а также проводя ДНК-исследование.
Немаловажным является и посещение невролога. Врач проведет первичный осмотр, по результатам которого выявит отличия болезни от других заболеваний.
К сожалению, в некоторых случаях осмотр у невролога не позволяет выявить характерные признаки заболевания из-за стертой клинической картины, поэтому врач может только заподозрить патологию, но поставить такой диагноз без более подробного обследования не получится.
Пациентов с подозрениями на данный недуг в обязательном порядке направляют на КТ и МРТ, а после чего врач-рентгенолог определит наличие изменений в головном мозге.
Важно понимать, что только обследование, проведенное на современных аппаратах, предоставит возможность врачам дать оценку возникшим изменениям в структуре головного мозга, на основании которых будет поставлен правильный диагноз.
Лечение и прогноз
К сожалению, в настоящее время заболевание считается неизлечимым и никакого специального лечения, направленного на остановку дегенеративных процессов пока разработать не удалось.
Поэтому все мероприятия направлены исключительно на снятие симптомов и улучшение качества жизни больного.
При своевременно начатом лечении, которое было подобрано верно, а также соблюдении всех рекомендаций врачей, больные получают все шансы на замедление дегенеративных процессов, происходящих на фоне протекания болезни.
Однако, полного выздоровления не наступает даже при правильной терапевтической тактике. Если имеет место быть синдром паркинсонизма, назначают агонисты дофамина, иногда пациентам показано применение амантадина. С целью снятия спастики выписываются лекарственные препараты с миорелаксирующим действием.
Как уже говорилось выше, почти всегда даже хорошее лечение не сможет остановить дальнейшее развитие заболевания, поэтому с течением времени у больных появляются новые симптомы патологии, что в конечном итоге приводит к смерти. Согласно официальным статистическим данным, после выявления заболевания до гибели больного проходит от 10 до 20 лет.
Продолжительность жизни имеет прямую связь с типом заболевания. Неблагоприятный прогноз имеет первый тип Масадо-Джозефа, больные с таким диагнозом, как правило, не живут дольше 10 лет после начала прогрессирования патологии.
Профилактические мероприятия сводятся к проведению пренатальной диагностики, а также в получении консультации у генетиков.
Если у ближайших родственников был диагностирован синдром Мачадо-Джозефа, важно со всей ответственностью подходить к планированию и рождению ребенка, так как генная мутация может передаться по наследству.
Многие генетики сходятся во мнении, что очень важно не допускать рождение ребенка, у которого имеется характерная генетическая мутация.
Источник: https://mrt-v-msk.ru/bolezn-machado-dzhozefa/





